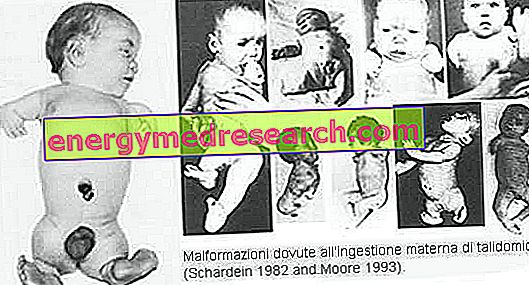
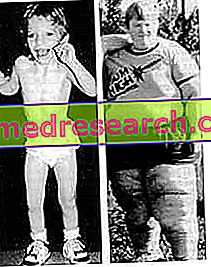
Свързани статии: Osteogenesis imperfecta дефиниция Несъвършена остеогенеза включва група от генетични заболявания, характеризиращи се с крехкост на костите и деформация на скелета при различни тежести. Понастоящем, въз основа на рентгенографски характеристики и молекулярно-генетичен анализ, се различават петнадесет различни типа. Основните форми на о