Какво е синдром на Марфан?
Синдромът на Марфан описва сложно наследствено нарушение на съединителната тъкан, което засяга главно очите, сърдечно-съдовата система и скелетната мускулна система. Въпреки това, като се има предвид, че всеки орган е съставен от съединителна тъкан, синдромът на Марфан може в най-добрия случай да унищожи и пречи на растежа и функцията на всеки анатомичен участък.
Синдромът се предава като автозомно доминантна черта: затова сме изправени пред тежко генетично заболяване с изключително променлива фенотипна експресия (дефектите могат да се различават значително от семейството до семейството или от пациента към пациента).
Това, което предизвиква синдрома на Марфан, е промяната на гена FBN1 (на хромозома 15), която кодира фибрилин-1, много важен гликопротеин на съединителя, който е структурната подкрепа за микрофибрилите.
Микрофибрили: състоящи се от фибрилин, микрофибрилите присъстват в извънклетъчната матрица, в която образуват препредаване за отлагане на еластин в еластичните влакна. Макар и вездесъщ в тялото, микрофибрилите изобилстват преди всичко в аортата, връзките и зоните на цилиарните тела (очното ниво).
Като автозомно доминантно разстройство, само децата, наследени от гена FBN-1, променени от двамата родители, са засегнати от синдрома на Марфан. Въпреки това, в един от 4 случая заболяването е резултат от спонтанни мутации при пациенти, които нямат фамилна анамнеза.
Името на заболяването идва от френския педиатър, който за пръв път я описа още през 1896 г. (А. Марфан), след което е необходимо да се изчака до 1991 г. да идентифицира променения ген, участващ в симптоматичната проява: откривателят е Ф. Рамирес.
Гледайте видеоклипа
X Гледайте видеоклипа в YouTubeПричини
Споменахме, че синдромът на Марфан е непосредственият израз на мутацията на ген, който кодира фибрилин-1. Опитваме се да задълбочим аргументацията, за да разберем кой механизъм е създаден, за да предизвика синдрома.
ФИБРИЛИН 1 е гликопротеинов компонент на еластина, който е от съществено значение за осигуряване и поддържане на еластичността и здравината на тъканите. При физиологични условия, фибрилин 1 се свързва с друг протеин, известен като TGF-бета (или трансформиращ фактор на растежа бета). Оказва се, че TGF-бета участва в вредни процеси на съдовия гладък мускул и извънклетъчния матрикс. Като се започне от тези предположения, някои автори смятат, че синдромът на Марфан се дължи, в допълнение към мутацията на гена FBN-1, също и на излишък на TGF-бета, особено в аортата, в сърдечните клапи и в белите дробове.
падане
Смята се, че синдромът на Марфан засяга 1 пациент на всеки 3 000-5 000 раждания и се проявява безразборно между мъже и жени, без да се пристрасти към точна раса. Статистиката показва, че 75% от пациентите имат положителна семейна история; при останалите 25% причината е в спорадични мутации, които изглежда са свързани по някакъв начин с напредналата възраст на бащата в момента на зачеването.
Децата с изключително тежки форми на синдрома на Марфан имат продължителност на живота по-малка от една година.
Преди еволюцията на стратегиите за открита операция на сърцето, повечето пациенти с синдром на Марфан имаха средна продължителност на живота от 32 години; благодарение на непрекъснатото подобряване на медицинските и фармакологичните терапии, в момента пациентите на синдрома на Марфан живеят средно до 60 години.
Признаци и симптоми
За задълбочаване: Синдром на Марфан
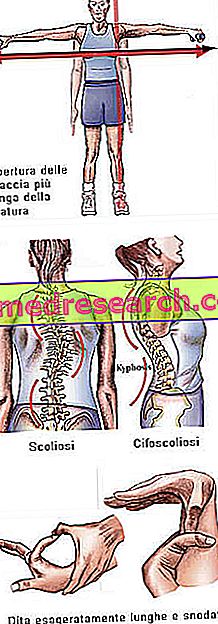
Синдромът на Марфан може да започне напълно безсимптомно. Засегнатите пациенти имат преувеличено дългосрочна структура и са непропорционално високи и слаби. Долните и горните крайници имат много по-голяма дължина от ствола (доликостеномегалия). Говорим и за арахнодактилите, за да изразим по-добре концепцията за прекомерната дължина на пръстите, типична за засегнатите от синдрома на Марфан: ръцете са сравнени с краката на паяка.
По отношение на височината, тези пациенти са къси със средна стойност над 97-ия персентил.
Сред другите отличителни черти, често срещани при пациенти с синдром на Марфан, също споменаваме:
- Отварянето на ръцете е по-голямо от височината
- Странични стави → преувеличена подвижност на ставите
- Деформация на гръдната стена
- Дислокация на лещата
- Горната част на тялото е по-слабо развита от долната
- Спонтанен пневмоторакс (11%)
- сколиоза
- Кожни стреси на бедрото, гърба, делтовидната, гръдната област
Сред най-проблематичните признаци, свързани със синдрома на Марфан, си спомняме пролапс на сърдечната клапа и недостатъчност на митралната клапа: подобно състояние може лесно да благоприятства дилатацията на аортния пръстен и дисекцията на аортата.
Таблицата показва признаците, които могат да бъдат открити при пациенти със синдром на Марфан. Описаните там герои не винаги присъстват, но голяма част от тях могат да бъдат намерени.
| Засегнатият анатомичен сайт | Възможни симптоми |
сладък | Striae в гръдната, лумбалната и сакралната област |
очи | Промяна на зрението, астигматизъм, отлепване на ретината, глаукома със затворен ъгъл, изкълчване на лещата, късогледство |
Структура на костите | Артралгия, кифосколиоза, долихостеномелия (прекомерна дължина на крайниците в сравнение с тялото), хипермобилност, високо небце, деформирани гърди, плоски стъпала, тесни и тесни китки, аномално повторно влизане / издатина на гръдната кост, сколиоза, извити рамене, спондилолистеза |
Дита | arachnodactyly |
бели дробове | Спонтанен пневмоторакс, диспнея, идиопатична белодробна обструктивна болест |
Промени в лицето | Ogal небце (малформация на небцето), мандибуларна ретрагенция (дефект на развитието на челюстта), удължено лице |
сърце | Ангина пекторис, аневризма на коремната аорта, сърдечна аритмия, дилатация / разкъсване / дисекция на гръдната аорта, аортна недостатъчност, пролапс на митралната клапа |
език | Трудност на езика |
диагноза
Като се имат предвид над 200 възможни мутации, използването на генетични маркери е почти невъзможно за диагностични цели.
Оценката на синдрома на Марфан не винаги е толкова непосредствена, тъй като фенотипната експресия на мутацията не винаги е очевидна и лесна за идентифициране. Диагностичното забавяне може сериозно да компрометира оцеляването на пациента: просто помислете за липсата на разпознаване на сърдечно-съдови проблеми.
Диагностичните критерии за синдрома на Марфан са разработени в международен план през 1996 г .: диагнозата се състои от изследване на семейната история, свързана с комбинация от основни и второстепенни показатели на синдрома.
Някои от многото използвани диагностични тестове са:
- ехокардиография
- магнитен резонанс и CT (за изследване на аортата)
- магнитно-резонансна ангиография (MRA) с контрастна течност (за да се видят вътрешните аортни структури)
- изследване с нарязани лампи (за анализ на възможното изместване на лещата)
- измерване на очното налягане (за да се подчертае възможното наличие на глаукома)
- генетични тестове (препоръчва се преди зачеване на дете, за да се установи или не синдромът)
терапии
Тъй като е генетично заболяване, няма лекарство или лечение, които да обърнат болестта.
Въпреки това, употребата на лекарства е от съществено значение за облекчаване на симптомите и за избягване на всякакви усложнения, особено сърдечни. За тази цел са особено показани лекарства за понижаване на кръвното налягане, като сартани (особено), АСЕ инхибитори и бета-блокери.
В контекста на синдрома на Марфан, пациентите, страдащи от сколиоза, могат също да следват специфични грижи, както и тези, засегнати от глаукома.
Хирургията е възможно да коригира аномалната дилатация на аортата, елемент, който често обединява по-голямата част от пациентите със синдром на Марфан.
Продължи: Синдром на Марфан - наркотици и грижи »


